Pathfinder – A Route Planner for Chemical Reaction Networks
Automated reaction network exploration enables the discovery of vast chemical space[1, 2, 3], consisting of many compounds highly interconnected via reactions. As a consequence, tracking what compounds are formed via which reaction sequence is a non-trivial task. While kinetic modeling provides the concentration or population development of a compound over time, a specific sequence of reactions resulting, for instance, in the most probable path towards a compound is not directly accessible. Furthermore, in a large and dense chemical reaction network containing a wide range of reaction barriers, kinetic modeling on time scales of interest might be too costly for the evaluation of populations in a network.[4]
This work aims to represent any chemical reaction network as a graph and analyze this graph to find paths, meaning sequences of reactions, connecting any two compounds in the network. An identified path should provide information on its feasibility of the reactions along the path with regard to availability of reagents and kinetic barriers. This yields a ranking of accessibility for all compounds in the network which can be employed to guide further explorations by focusing on compounds with high accessibility according to Pathfinder. Such a guiding approach tames the combinatorial explosion of an exploration and gives the explorer more control over a running exploration. In addition, for economic kinetic modeling, the network can be pruned based on the ranking of compounds, again starting by only including compounds with high accessibility.
We employ such a graph-based approach to build and analyze a chemical reaction network of the disproportionation of iodine with water and improve explorations based on the results within our SCINE framework.[5]
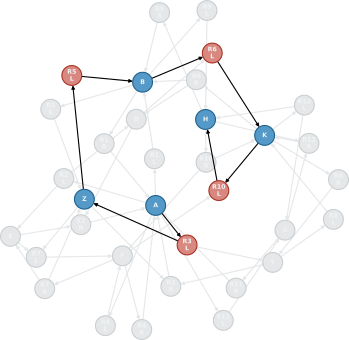
Figure 1: Schematic representation of a path from compound A to compound H in an arbitrary chemical reaction network.
[1] G. N. Simm, A. C. Vaucher, M. Reiher, J. Phys. Chem. A, 2019, 123 (2), 385–399.
[2] J. P. Unsleber, M. Reiher, Annu. Rev. Phys. Chem., 2020, 71 (1), 121–142.
[3] J. P. Unsleber, S. A. Grimmel, M. Reiher, arXiv:2202.13011 [physics.chem-ph], 2022.
[4] J. Proppe, M. Reiher, J. Chem. Theory Comput., 2019, 15 (1), 357–370.
[5] SCINE - Software for Chemical Interaction Networks. https://scine.ethz.ch/ (accessed February 24, 2022).