Search for long-lasting electronic coherence using on-the-fly ab initio semiclassical dynamics
Using a combination of high-level ab initio electronic structure methods with efficient on-the-fly semiclassical evaluation of nuclear dynamics, we performed a massive scan of small polyatomic molecules searching for a long-lasting oscillatory dynamics of the electron density triggered by the outer-valence ionization. We observed that in most of the studied molecules, either the sudden removal of an electron from the system does not lead to the appearance of the electronic coherence or the created coherences become damped by the nuclear rearrangement on a time scale of a few femtoseconds. However, we report several so far unexplored molecules with the electronic coherences lasting up to 10 fs, which can be good candidates for experimental studies. In addition, we present the full-dimensional simulations of the electronic coherences coupled to nuclear motion in several molecules which were studied previously only in the fixed nuclei approximation.
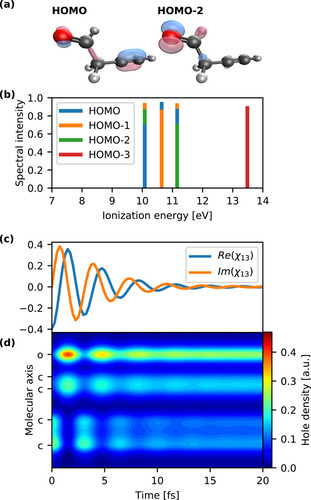
FIG. 1. Ionization spectrum and the coupled electron–nuclear dynamics triggered by the ionization out of the HOMO of the but-3-ynal molecule. (a) Molecular orbitals involved in the hole-mixing. (b) First four computed cationic states. (c) Time evolution of the electronic coherence between the first and the third cationic states created after removal of the HOMO electron. Dynamics was performed with the semiclassical on-the-fly thawed Gaussian approximation. (d) Time evolution of the hole density along the molecular axis. The charge initially localized in the HOMO orbital migrates back and forth between the alkyne and aldehyde moieties of the molecule before being trapped by the nuclear motion.